Ultrarzadka choroba dziedziczna – czy ją znasz?
Czego dowiesz się z artykułu:
- Co jest istotą choroby Huntera i jakie są jej charakterystyczne objawy
- Dlaczego rola laryngologa w diagnostyce i leczeniu jest taka ważna
- Jakie mamy dostępne terapie
- Gdzie skierować pacjenta z podejrzeniem choroby.
Choroba Huntera [ICD – E 76.1, OMIM – 309900] to ultrarzadka choroba dziedziczna. Cechy charakterystyczne: karłowatość, grube rysy twarzy, powiększenie wątroby i śledziony, przykurcze palców, opóźnienie umysłowe i głuchota. Zespół chorobowy został opisany po raz pierwszy u dwóch braci przez dr. Charlesa A. Huntera w 1917 r. (Charles A. Hunter (1873–1955)) [1].
|
Choroby rzadkie – częstość w populacji nie jest wyższa niż 1 na 2000 osób lub rzadziej
Choroby ultrarzadkie - występują z częstością 1 na 50 tysięcy lub mniejszą
|
Choroba Huntera
Etiopatogeneza
Choroba Huntera, mukopolisacharydoza typu II [MPS II] należy do wrodzonych defektów metabolicznych wywołanych zaburzeniami metabolizmu glikozaminoglikanów (GAG) – kwaśnych mukopolisacharydów. Mukopolisacharydozy (MPS) to heterogenna grupa dziedzicznych zaburzeń metabolicznych, z których każde wiąże się z niedoborem co najmniej jednego z enzymów biorących udział w katabolizmie glikozaminoglikanów (GAG). Prawidłowe GAG są podstawową i niezwykle istotną częścią składową tkanki łącznej, składają się z łańcuchów cząsteczek cukrów i podlegają rozkładowi w lizosomach dzięki działaniu zestawu właściwych enzymów. Brak lub niedobór enzymu powoduje zaburzenie procesu katabolizmu GAG w lizosomach, co prowadzi do nieprawidłowych, patologicznych form metabolizmu, w konsekwencji skutkując nieprawidłową budową i funkcją wszystkich elementów tkanki łącznej. Nieprawidłowy metabolizm GAG powoduje coraz rozleglejsze skutki w organizmie chorego, w związku z czym objawy przedmiotowe i podmiotowe choroby nasilają się wraz z wiekiem. Opisywanych jest siedem typów MPS (I, II, III, IV, VI, VII i IX) ze znanymi niedoborami 11 różnego typu enzymów.

Glikozaminoglikany (GAG) dawniej nazywane mukopolisacharydami są polimerami polianionowymi, z których większość zawiera reszty węglowodanowe: N-asctyloheksozoaminę i kwas uronowy. |
Istotą choroby Huntera jest niedobór aktywności lizosomalnego enzymu sulfatazy siarczano-iduronowej, co powoduje patologiczną zmianę metabolizmu siarczanu dermatanu i heparanu w tkankach organizmu chorego z następowymi zaburzeniami metabolizmu tkanki łącznej całego organizmu oraz nadmiernym wydalaniem tych związków z moczem [2, 3].
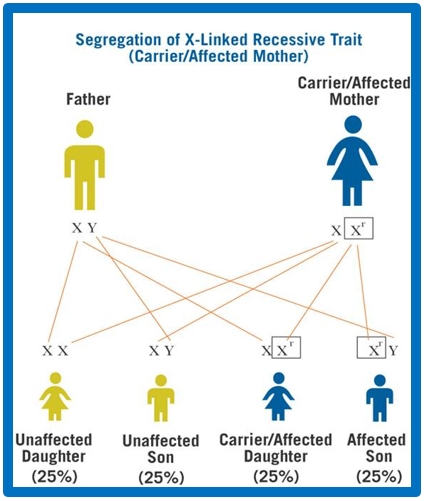
Dziedziczenie
Dziedziczenie ma charakter recesywny związany z chromosomem X (w odróżnieniu od innych postaci mukopolisacharydoz, które są dziedziczone autosomalnie recesywnie).
Gen IDS znajduje się na długim ramieniu chromosomu X (Xq28), nosicielami są kobiety, mężczyźni chorujący na MPS II niezwykle rzadko mają potomstwo.
Pełna treść dotępna tylko dla zalogowanych użytkowników.